Diagnose scale issue of summary statistics#
suppressPackageStartupMessages(library(tidyverse))
library(data.table)
library(ggplot2)
library(patchwork)
In an association of a single SNP \(j\), we have a simple linear model (Yang, Jian, et al(2012)):
Based on OLS, we have
and
Based on ANOVA, we have
Based on probability, we have Z-score
Reformatting Eq(4), we derive
By combining Eq(3) and Eq(5) into Eq(6), we have
Here, we assume trait phenotype \(y\) is standardized (thus, \(y^Ty = n\)) and \(D_j = 2p_j(1-p_j)n\). If only summary statistics Z score is provided, we can re-estimate \(\hat{b}_j\) and \(\sigma_b\) given allele frequency \(p\), sample size \(n\) and zscore \(z\), therefore,
Also, we have
############################################
### Run a simulation to simulate gene exression
### y = beta * x + e, where y is scaled gene expression
### x is the genotype at 0/1/2 scale, BETA and SE
### is the original effect size and standard error
### N is the sample size
### A1Freq is the allele frequency
### zz is the zscore
############################################
set.seed(1234)
sumPath = "~/Documents/personal/SbayesAnno/downSampling/Verify0319/summary/"
smrIndFileSuffix = "ukbg-chr22-causal-model-50k-ind"
geneLDFile = paste0(sumPath,smrIndFileSuffix)
xqtl = fread(paste0(geneLDFile,".q.query.gz"))
test = xqtl %>% mutate(zz = BETA/SE) %>%
mutate(beta = zz / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2)),
betaScale = BETA * sqrt(2 * A1Freq * (1-A1Freq)),
se = 1 / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2))
)
head(test)
options(repr.plot.width = 12, repr.plot.height = 6)
#### beta based on zscore and Original beta
p1 = ggplot(test, aes(x = beta, y = BETA,)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
y = "Orignal Beta",
x = "Beta based on Zscore",
title = "Fig 1.1 Orignal Beta vs Beta based on Zscore in the simulation"
) + theme_minimal();
#### beta based on zscore and Original beta
p2 = ggplot(test, aes(x = beta, y = betaScale)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
x = "Beta based on Zscore",
y = "scaled Beta",
title = "Fig 1.2 scaled Beta vs Beta based on Zscore in the simulation"
) + theme_minimal();
(p1 + p2)
print(paste0("correlation between beta and BETA is ", cor(test$beta, test$BETA)))
print(paste0("correlation between beta and betaScale is ", cor(test$beta, test$betaScale)))
| GeneID | GeneChr | GeneLength | GenePhyPos | SNPID | SNPChr | SNPPhyPos | A1 | A2 | A1Freq | BETA | SE | Pvalue | N | zz | beta | betaScale | se |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <int> | <int> | <dbl> | <chr> | <int> | <int> | <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | <dbl> | <dbl> |
| ENSG00000187905 | 22 | -999 | 21409346 | rs428214 | 22 | 21361060 | T | G | 0.155100 | -0.006123455 | 0.008735706 | 4.833227e-01 | 50000 | -0.7009685 | -0.006123393 | -0.003134874 | 0.008735618 |
| ENSG00000187905 | 22 | -999 | 21409346 | rs373092 | 22 | 21361870 | G | T | 0.027860 | -0.511740506 | 0.019078813 | 2.225000e-308 | 50000 | -26.8224492 | -0.511735459 | -0.119102192 | 0.019078625 |
| ENSG00000187905 | 22 | -999 | 21409346 | rs398577 | 22 | 21362849 | A | C | 0.139213 | -0.275573105 | 0.009060138 | 2.225000e-308 | 50000 | -30.4159945 | -0.275319807 | -0.134908581 | 0.009051810 |
| ENSG00000187905 | 22 | -999 | 21409346 | rs2075274 | 22 | 21363306 | A | G | 0.371860 | -0.301651835 | 0.006405235 | 2.225000e-308 | 50000 | -47.0945754 | -0.301531294 | -0.206176388 | 0.006402676 |
| ENSG00000187905 | 22 | -999 | 21409346 | rs2516542 | 22 | 21363505 | T | C | 0.126070 | -0.342185229 | 0.009403473 | 2.225000e-308 | 50000 | -36.3892402 | -0.342181893 | -0.160627706 | 0.009403381 |
| ENSG00000187905 | 22 | -999 | 21409346 | rs2075276 | 22 | 21363744 | C | T | 0.498304 | -0.434683323 | 0.006030275 | 2.225000e-308 | 50000 | -72.0834933 | -0.433913672 | -0.307365757 | 0.006019598 |
[1] "correlation between beta and BETA is 0.999999335847599"
[1] "correlation between beta and betaScale is 0.957713529698195"
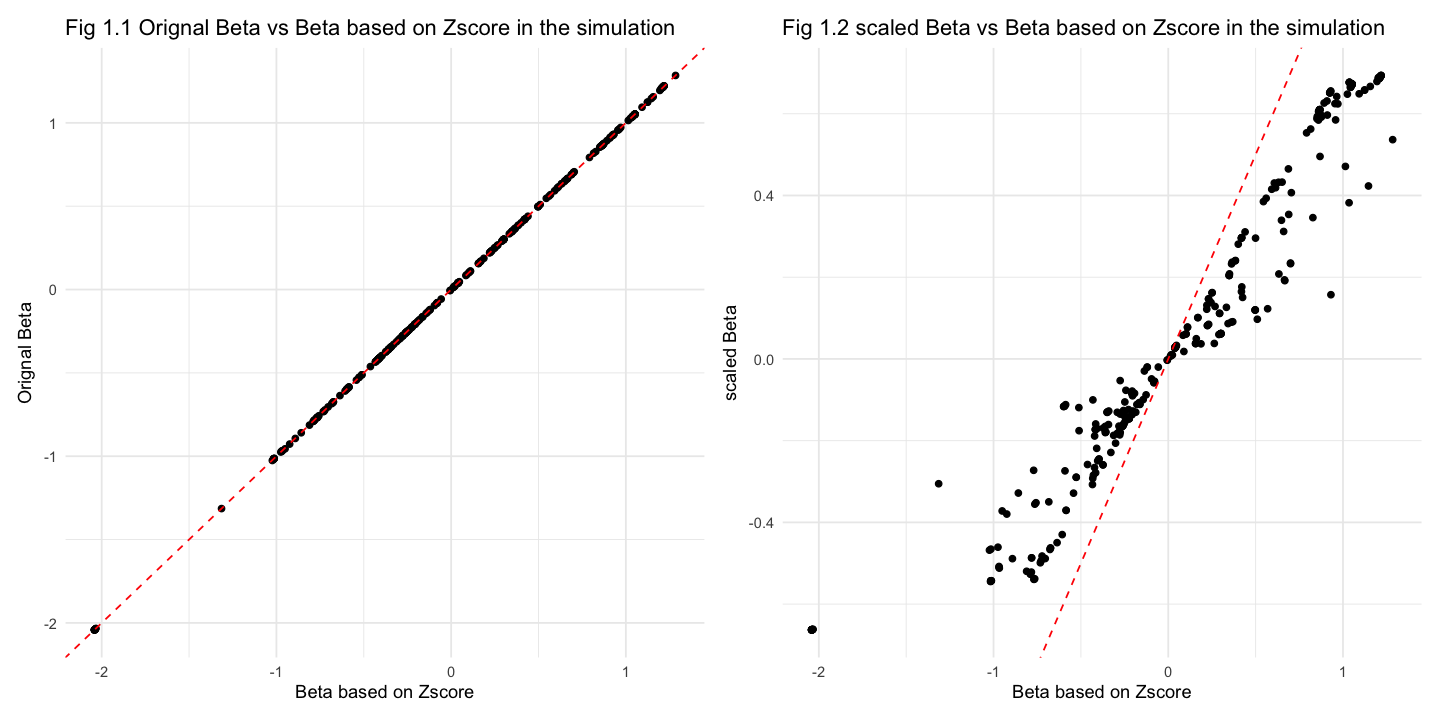
Fig 1.1 suggests that the original beta values are very well captured by the Z-score-based estimates. In Fig 1.2 (right plot), the data points systematically deviate from the reference line \(y = x\), indicating a upward biased (Scaled Beta is larger than Beta based on Z-score).
############################################
### blood pQTLs
############################################
library(data.table)
library(tidyverse)
beqtlPath = "~/Downloads/data/xqtl/"
beqtl = fread(paste0(beqtlPath,"ukb-ppp-pGWAS-v1-eur-hg19-pro3k-chr22-cis1M-HM3q.query.gz"))
test = beqtl %>% mutate(zz = BETA/SE) %>%
mutate(beta = zz / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2)),
betaScale = BETA * sqrt(2 * A1Freq * (1-A1Freq)),
se = 1 / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2))
) %>% slice_sample(n = 10000)
head(test)
options(repr.plot.width = 12, repr.plot.height = 6)
p1 = ggplot(test, aes(x = beta, y = BETA)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
y = "Beta from raw data",
x = "Beta based on Zscore",
title = "Randomly selected 10,000 SNPs from genes for blood pQTLs"
) + theme_minimal()
p2 = test %>%
mutate( betaScale = BETA * sqrt(2 * A1Freq * (1-A1Freq))) %>%
ggplot( aes(x = beta, y = betaScale)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
y = "Fig 2.1 Beta based on Zscore adjusted by snp2pq (beta * sqrt(2 * A1Freq * (1-A1Freq)))",
x = "Fig 2.2 Beta based on Zscore",
title = "Randomly selected 10,000 SNPs from genes for blood pQTLs"
) + theme_minimal()
p1 + p2
print(paste0("correlation between beta and BETA is ", cor(test$beta, test$BETA)))
print(paste0("correlation between beta and betaScale is ", cor(test$beta, test$betaScale)))
| GeneID | GeneChr | GeneLength | GenePhyPos | SNPID | SNPChr | SNPPhyPos | A1 | A2 | A1Freq | BETA | SE | Pvalue | N | zz | beta | betaScale | se |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <int> | <int> | <dbl> | <chr> | <int> | <int> | <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | <dbl> | <dbl> |
| OID20838_ENSG00000100083 | 22 | -999 | 37621144 | rs86582 | 22 | 37603390 | T | C | 0.194599 | 0.013366000 | 0.00972440 | 0.169292502 | 32922 | 1.3744807 | 0.0135300154 | 0.0074832945 | 0.009843729 |
| OID30372_ENSG00000099977 | 22 | -999 | 23975534 | rs17004053 | 22 | 24250835 | G | A | 0.032967 | -0.091722200 | 0.02052610 | 0.000007875 | 33924 | -4.4685644 | -0.0960545088 | -0.0231606009 | 0.021495608 |
| OID30382_ENSG00000100345 | 22 | -999 | 36334645 | rs5999726 | 22 | 35502070 | A | C | 0.620594 | 0.012363900 | 0.00778328 | 0.112168685 | 33924 | 1.5885205 | 0.0125677829 | 0.0084845022 | 0.007911628 |
| OID20865_ENSG00000130638 | 22 | -999 | 45758552 | rs1555208 | 22 | 46577210 | T | G | 0.090022 | -0.004931240 | 0.01465610 | 0.736521511 | 32922 | -0.3364633 | -0.0045813734 | -0.0019960017 | 0.013616264 |
| OID30352_ENSG00000100024 | 22 | -999 | 24511248 | rs9608196 | 22 | 24163081 | C | T | 0.116033 | 0.000867572 | 0.01156900 | 0.940221796 | 33942 | 0.0749911 | 0.0008987183 | 0.0003929428 | 0.011984333 |
| OID30708_ENSG00000100342 | 22 | -999 | 36260300 | rs2481 | 22 | 36677400 | A | G | 0.132781 | 0.009543770 | 0.01004600 | 0.342108712 | 33985 | 0.9500070 | 0.0107383219 | 0.0045800194 | 0.011303414 |
[1] "correlation between beta and BETA is 0.997710343811819"
[1] "correlation between beta and betaScale is 0.921397747836"
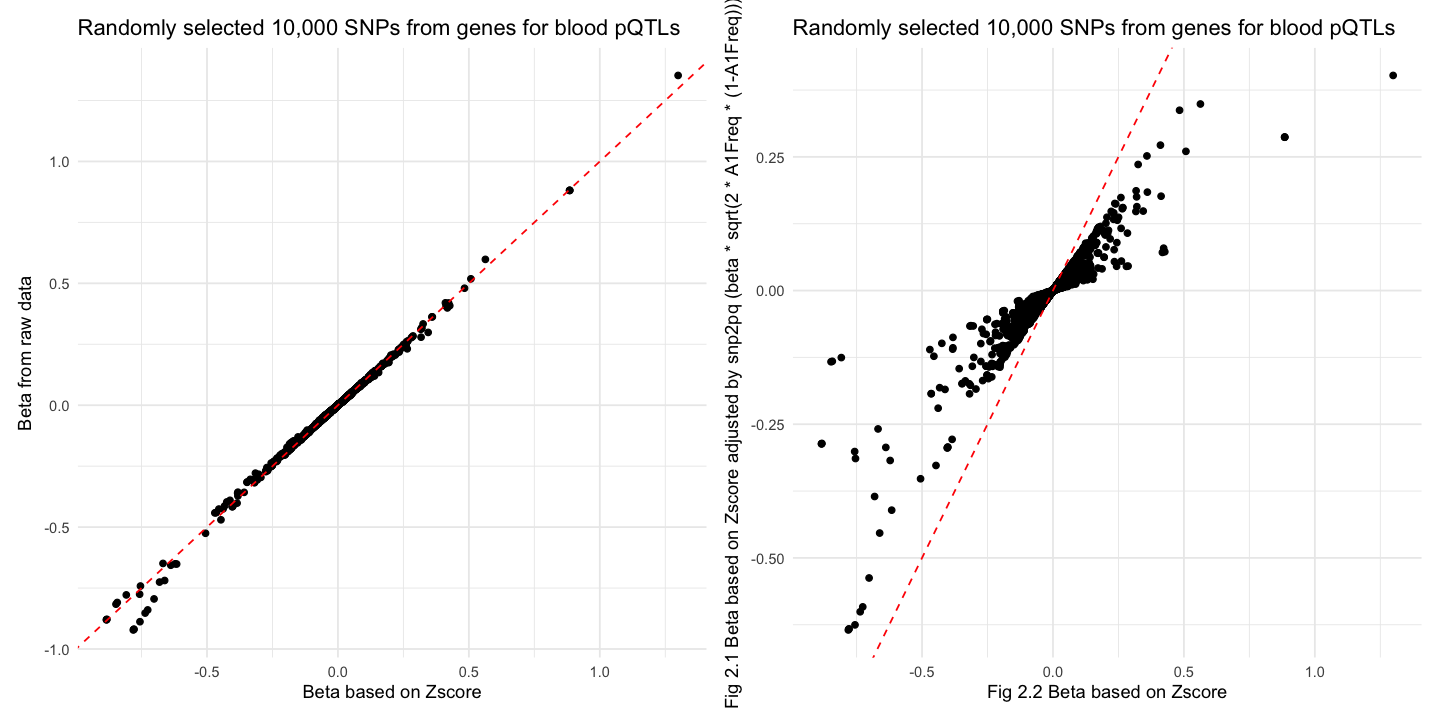
The transformation behavior in blood pQTLs data is highly similar to the simulated results. The systematic bias pattern (negative values shrinking, positive values stretching) remains evident.
############################################
### Brain eQTLs
############################################
library(data.table)
library(tidyverse)
beqtlPath = "~/Downloads/data/xqtl/"
beqtl = fread(paste0(beqtlPath,"QC-HM3-brain-gene16620-eqtl-1086269-cis1M-chr1-22-2025-03-17.query.gz"))
test = beqtl %>% mutate(zz = BETA/SE) %>%
mutate(beta = zz / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2)),
betaScale = BETA * sqrt(2 * A1Freq * (1-A1Freq)),
se = 1 / sqrt(2 * A1Freq * (1-A1Freq) * (N-1 + zz^2))
) %>% slice_sample(n = 10000)
head(test)
options(repr.plot.width = 12, repr.plot.height = 6)
p1 = ggplot(test, aes(x = beta, y = BETA)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
y = "Beta from raw data",
x = "Beta based on Zscore",
title = "Randomly selected 10,000 SNPs from genes for brain eQTLs"
) + theme_minimal()
p2 = test %>%
mutate( betaScale = BETA * sqrt(2 * A1Freq * (1-A1Freq))) %>%
ggplot( aes(x = beta, y = betaScale)) +
geom_point() +
geom_abline(intercept = 0, slope = 1, linetype = "dashed", color = "red") +
labs(
y = "Fig 3.1 Beta based on Zscore adjusted by snp2pq (beta * sqrt(2 * A1Freq * (1-A1Freq)))",
x = "Fig 3.2 Beta based on Zscore",
title = "Randomly selected 10,000 SNPs from genes for brain eQTLs"
) + theme_minimal()
p1 + p2
print(paste0("correlation between beta and BETA is ", cor(test$beta, test$BETA)))
print(paste0("correlation between beta and betaScale is ", cor(test$beta, test$betaScale)))
| GeneID | GeneChr | GeneLength | GenePhyPos | SNPID | SNPChr | SNPPhyPos | A1 | A2 | A1Freq | BETA | SE | Pvalue | N | zz | beta | betaScale | se |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| <chr> | <int> | <int> | <int> | <chr> | <int> | <int> | <chr> | <chr> | <dbl> | <dbl> | <dbl> | <dbl> | <int> | <dbl> | <dbl> | <dbl> | <dbl> |
| ENSG00000143771.12 | 1 | -999 | 224555860 | rs4653593 | 1 | 224881970 | A | G | 0.213115 | -0.006912926 | 0.03710873 | 0.85221859 | 2865 | -0.1862884 | -0.006010617 | -0.004003497 | 0.03226512 |
| ENSG00000146909.8 | 7 | -999 | 156754138 | rs4078489 | 7 | 155858530 | C | T | 0.371585 | -0.006239648 | 0.03607818 | 0.86269234 | 2865 | -0.1729480 | -0.004728891 | -0.004264100 | 0.02734286 |
| ENSG00000254343.2 | 8 | -999 | 121066430 | rs10093792 | 8 | 121515110 | A | G | 0.314208 | -0.097312212 | 0.03977902 | 0.01443229 | 2865 | -2.4463199 | -0.069559050 | -0.063883274 | 0.02843416 |
| ENSG00000272512.1 | 1 | -999 | 932388 | rs2250833 | 1 | 1854321 | A | G | 0.306011 | 0.044837367 | 0.03829468 | 0.24165865 | 2865 | 1.1708511 | 0.033562277 | 0.029221325 | 0.02866486 |
| ENSG00000213199.8 | 7 | -999 | 150747611 | rs11984138 | 7 | 150300735 | A | G | 0.224044 | -0.004591339 | 0.03181196 | 0.88524191 | 2865 | -0.1443274 | -0.004573626 | -0.002707320 | 0.03168923 |
| ENSG00000139209.16 | 12 | -999 | 47192368 | rs2465230 | 12 | 47324596 | T | C | 0.423497 | -0.087216511 | 0.03631631 | 0.01632444 | 2865 | -2.4015800 | -0.064155329 | -0.060945222 | 0.02671380 |
[1] "correlation between beta and BETA is 0.978783611798265"
[1] "correlation between beta and betaScale is 0.931821297460271"
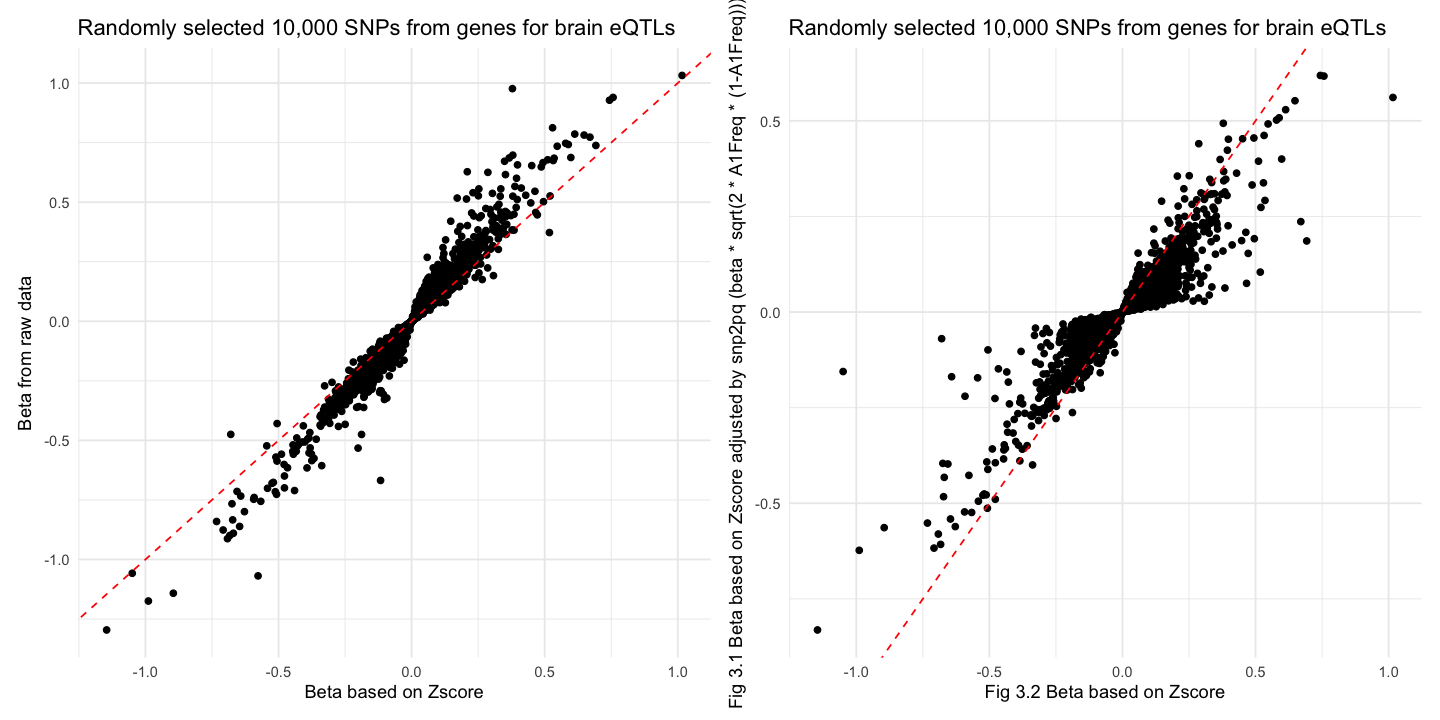
Similar trend was found in brain eQTLs but Fig 3.1 had larger variance since here we use unique total sample size for all eQTLs.
# Extreme effect for snp rs7749880
# /QRISdata/Q4062/softwares/smr/smr --beqtl-summary BrainMeta_cis_eQTL_chr6 --snp rs7749880 --out myquery --query
# SNP Chr BP A1 A2 Freq Probe Probe_Chr Probe_bp Gene Orientation b SE p
# rs7749880 6 58346584 A G 0.469945 ENSG00000271761.1 6 58229156 AL021368.1 - -0.137552 0.0222519 6.34526e-10
# rs7749880 6 58346584 A G 0.469945 ENSG00000215190.9 6 58280066 LINC00680 - **-3156.06** 2.8288 0
# Cis-sQTL mapping was conducted using FastQTL, testing for associations between genetic variants located within ±1 Mb of
# each gene’s transcription start site (TSS). The analysis incorporated the covariates detailed in Section 4.1, along with
# 15 PEER factors derived from splicing quantifications (Section 3.5.2) (GTEx Consortium, 2020).
# FastQTL(Ongen, Halit, et al.(2016)) utilizes normalized gene expression data, covariate matrices, and dosage genotype information
# within a regression framework to identify statistically significant associations between genetic variants and molecular phenotypes,
# thereby uncovering both cis (proximal) and trans (distal) QTLs.