Simulation#
Schematic overview of various simulation settings
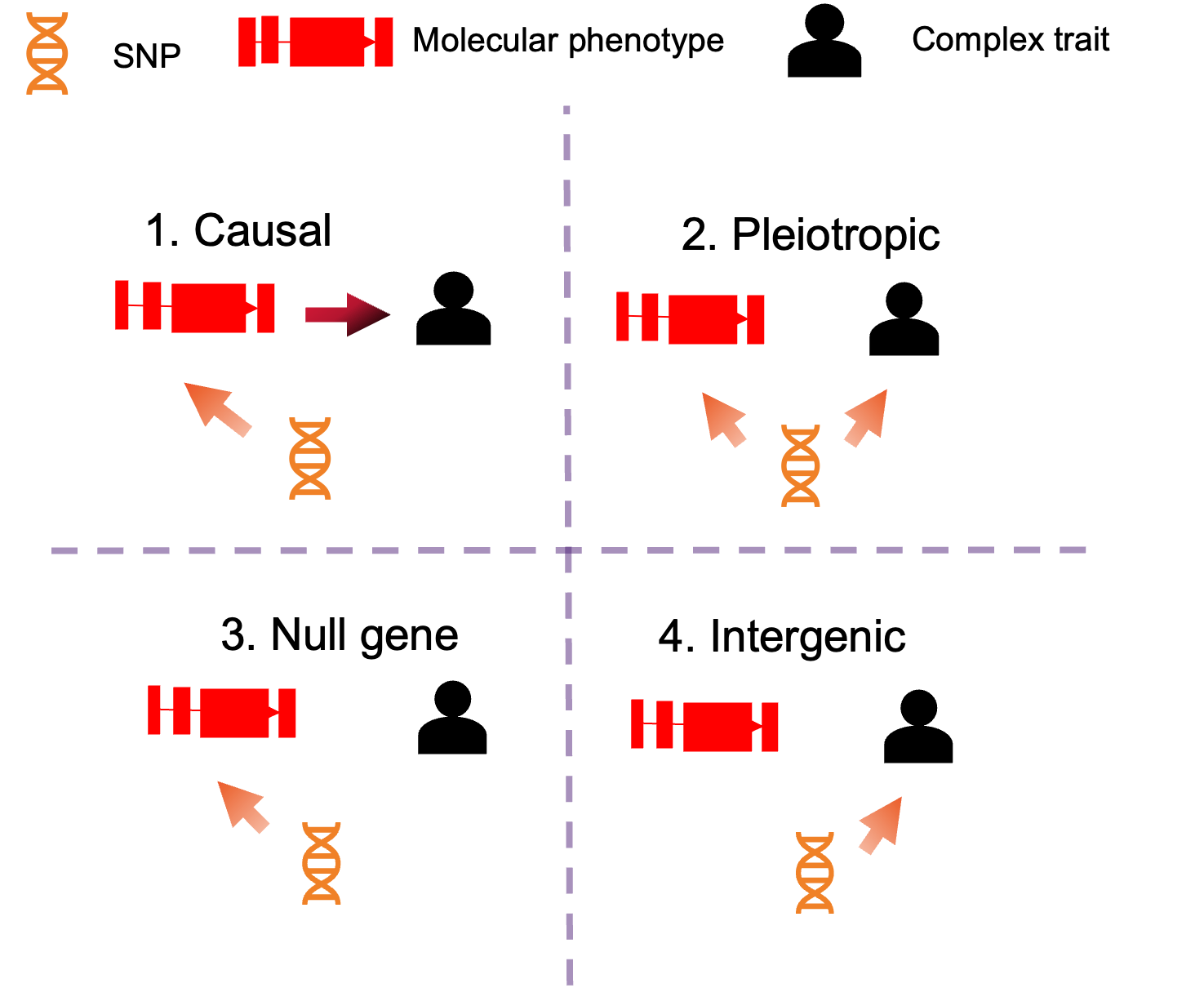
R scripts#
rm(list = ls())
library(SBayesOmics)
datPathInPack <- system.file("extdata/pseudo1kgchr22", package = "SBayesOmics")
trainBfilePrefix <- "psuedo-1kg-chr22-causal-model-ind-10k-snp-11555"
paramSuffix = "1kg-cis-50k-"
trainBfile <- paste0(datPathInPack,"/",trainBfilePrefix)
snp2ldFile <- paste0(paramSuffix,"sub-all-block-chr22-ld-2-snp-map.rds")
WithoutLDAllSNP2GeneFile <- paste0(paramSuffix,"sub-all-block-","chr22-snp-2-gene-map.rds")
mapSnpGeneAcrossChrFile <- paste0(datPathInPack,"/",WithoutLDAllSNP2GeneFile)
mapLd2SnpFile <- paste0(datPathInPack,"/",snp2ldFile)
sim <- simGWASMainOverlap(isIndLevelBool = TRUE,
seed = 1,
trainBfile,
mapSnpGeneAcrossChrFile,
mapLd2SnpFile,
simModel = "b", ## a: causal model only; b: pleitropic model only; c: intergenic model only
geneOverlap = "c", ## random overlap
traitType = "a", ## continuous traits
indNum = 2000, ## gwas number
NumCGCau = 10, ## number of causal genes
NumCVPerGCau = 10, ### number of causal variants per gene
NumCGPle = 10, ## number of causal pleiotropic genes;
NumCVPerGPle = 10, ## number of causal variants per pleiotropic gene
NumCGNull = 10, ### number of null gene
NumCVPerGNull = 10, ### number of causal variants per null gene
NumCVIG = 10, ## number of causal variants in intergenic region
realGeno = TRUE,
ldwBool = TRUE,
cisOnly = TRUE,
h2cis = 0.5, ## cis-heritability
h2snp = 0.1, ## total snp heritability
h2med = 0.05, ## mediated heritability
outPath = "",
smrPath = "" ,
totalOverlapBool = FALSE,
smrIndGenePath = "",
smrIndFileSuffix = "mac",
cauPleWithinGene = FALSE
)